سندرم آلپورت

سندرم آلپورت
سندرم آلپورت یک بیماری ژنتیکی است که در آن کلیه ها پروتئین های کلاژن نوع IV طبیعی تولید نمیکنند. کلاژن نوع IV از سه زنجیره کلاژن مجزا (زنجیره های آلفا) تشکیل شده است که مانند طناب به هم میپیچند. این زنجیره ها آلفا ۳، آلفا ۴ و آلفا ۵ هستند. اگر بدن یکی از این زنجیره ها را تولید نکند، دو زنجیره دیگر نمی توانند با هم ترکیب شوند. این منجر به شدیدترین علائم سندرم آلپورت می شود.
اگر بدن تمام زنجیره ها را تولید کند، اما یکی را به درستی نسازد، گاهی اوقات زنجیرهها نمیتوانند با هم ترکیب شوند و گاهی اوقات زنجیرهها با هم ترکیب میشوند اما به درستی عمل نمیکنند. علائم سندرم آلپورت ممکن است در این موارد خفیفتر باشد.
کلاژن نوع IV یک پروتئین مهم در غشاهای فیلتراسیون در کلیهها (غشاهای پایه گلومرولی یا GBM) است.
GBM بخشی از یک ساختار سه لایه است که خون را فیلتر میکند تا سموم و سایر محتویاتی را که بدن برای تولید ادرار نیازی به آنها ندارد، حذف کند. GBM همچنین اطمینان حاصل میکند که محتویاتی مانند سلول های خونی و پروتئین ها به جای ورود به ادرار، در خون باقی می مانند.
وقتی GBM به درستی عمل نکند، خون یا پروتئین ها می توانند به ادرار نشت کنند. توانایی کلیه ها برای فیلتر کردن ادرار نیز به مرور زمان بدتر می شود که خطر نارسایی کامل کلیه را افزایش می دهد.
کلاژن نوع IV نیز در گوش ها و چشم ها وجود دارد. بنابراین در صورت داشتن سندرم آلپورت، این بیماری می تواند باعث ایجاد مشکلاتی در بینایی و شنوایی و همچنین کلیهها شود.
وراثت سندرم آلپورت چگونه است؟
سندرم آلپورت به سه شکل ژنتیکی مختلف منتقل می شود:
1. سندرم آلپورت وابسته به کروموزوم X یا (XLAS)
در این نوع، ژن معیوب مربوط به تولید زنجیره آلفا ۵ (COL4A5) بر روی کروموزوم X قرار دارد؛ یکی از دو کروموزوم جنسی (X و Y).
مردان تنها یک کروموزوم X دارند، بنابراین اگر این کروموزوم دچار جهش شود، احتمال بروز علائم شدید بیشتر است.
زنان دارای دو کروموزوم X هستند. اگر یکی از آنها جهشیافته باشد، معمولاً علائم خفیفتری نشان میدهند، چون کروموزوم سالم تا حدی جبران میکند.
از نظر وراثت:
مردان کروموزوم Y را به پسران و کروموزوم X را به دختران خود منتقل میکنند؛ بنابراین نمی توانند XLAS را به پسران خود انتقال دهند، اما تمام دختران آنها ناقل خواهند بود.
زنان میتوانند هر یک از دو کروموزوم X خود را به فرزندانشان منتقل کنند، در نتیجه ۵۰٪ احتمال دارد بیماری را به هر یک از فرزندان (دختر یا پسر) انتقال دهند.
XLAS شایعترین نوع سندرم آلپورت است و حدود 60 تا 80 درصد موارد را شامل میشود.
2. سندرم آلپورت با الگوی اتوزومال مغلوب (ARAS)
در این نوع، ژن های COL4A3 و COL4A4 که پروتئین های آلفا ۳ و آلفا ۴ را تولید می کنند، روی کروموزوم ۲ قرار دارند.
الگوی “اتوزومال مغلوب” به این معناست که برای بروز بیماری، باید هر دو نسخه از ژن (یکی از پدر و یکی از مادر) دچار جهش باشند.
والدینی که فقط یک نسخه از ژن معیوب را دارند، معمولاً هیچ علامتی ندارند و از ناقل بودن خود آگاه نیستند.
در صورت ناقل بودن هر دو والد:
احتمال ابتلای فرزند به سندرم آلپورت ۲۵ درصد است (اگر هر دو ژن معیوب را به ارث ببرد).
۵۰ درصد احتمال دارد که کودک تنها ناقل باشد (بدون علائم)
۲۵ درصد نیز احتمال دارد که ژن های سالم را از هر دو والد به ارث ببرد.
ARAS به جنسیت فرد وابسته نیست و شدت علائم آن در مردان و زنان یکسان است.
3. سندرم آلپورت با الگوی اتوزومال غالب (ADAS)
در الگوی اتوزومال غالب، تنها وجود یک نسخه جهش یافته از یک ژن برای بروز بیماری کافی است.
در نوع ADAS از سندرم آلپورت، جهش در یکی از ژن های COL4A3 یا COL4A4 که روی کروموزوم ۲ قرار دارند، باعث بروز بیماری می شود. این ژن ها مسئول تولید پروتئین هایی هستند که در ساختار غشای پایه کلیه، گوش و چشم نقش دارند.
ویژگی های ADAS:
این نوع از بیماری به جنسیت وابسته نیست؛ به همین دلیل، احتمال بروز علائم و شدت آن در زنان و مردان یکسان است.
فرد مبتلا به ADAS دارای 50 درصد احتمال انتقال ژن معیوب به هر یک از فرزندان خود است. در صورت انتقال، فرزند نیز به ADAS مبتلا خواهد شد.
این نوع حدود 25 تا 35 درصد از موارد سندرم آلپورت را شامل می شود.
سندرم آلپورت چه کسانی را تحت تأثیر قرار می دهد؟
سندرم آلپورت میتواند هر کسی را تحت تأثیر قرار دهد، زیرا یک بیماری ژنتیکی است. با این حال، در حدود 15% موارد، میتواند زمانی ایجاد شود که هیچ یک از والدین ژن جهش یافته را نداشته باشند.
علائم و علل سندرم آلپورت
سندرم آلپورت چگونه باعث نارسایی کلیه می شود؟
در سندرم آلپورت، غشای پایه گلومرولی (GBM) که مسئول فیلتر کردن خون در کلیههاست، عملکرد طبیعی خود را از دست میدهد. در نتیجه، موادی مانند خون و پروتئین که نباید وارد ادرار شوند، از این فیلتر عبور کرده و در ادرار ظاهر می شوند.
علاوه بر این، GBM توانایی لازم برای حمایت از سلول هایی که در دو سوی آن قرار دارند را ندارد. این ضعف ساختاری، موجب تحریک و التهاب سلولهای اطراف میشود.
پودوسیتها، سلول هایی هستند که وظیفه تولید کلاژن نوع IV و ساخت GBM را بر عهده دارند. زمانی که محیط اطراف این سلولها ملتهب می شود، آنها سعی می کنند با تولید بیشتر کلاژن IV به ترمیم GBM کمک کنند. اما این تلاش منجر به ضخیم و نامنظم شدن GBM می شود.
این تغییرات ساختاری در GBM باعث می شود که نشت پروتئین به ادرار (پروتئینوری) افزایش یابد، که یکی از نشانه های کلیدی بیماری کلیوی در سندرم آلپورت است.
با گذشت زمان، با نشت بیشتر پروتئین به ادرار و ضخیمتر شدن GBMها، ممکن است بافت اسکار (فیبروز) ایجاد شود. در نتیجه، توانایی کلیهها برای فیلتر کردن خون کاهش می یابد که باعث بیماری مزمن کلیه می شود. با ایجاد بافت اسکار بیشتر، عملکرد کلیه بدتر می شود تا اینکه در نهایت کلیهها از کار میافتند و نارسایی کلیه رخ می دهد.
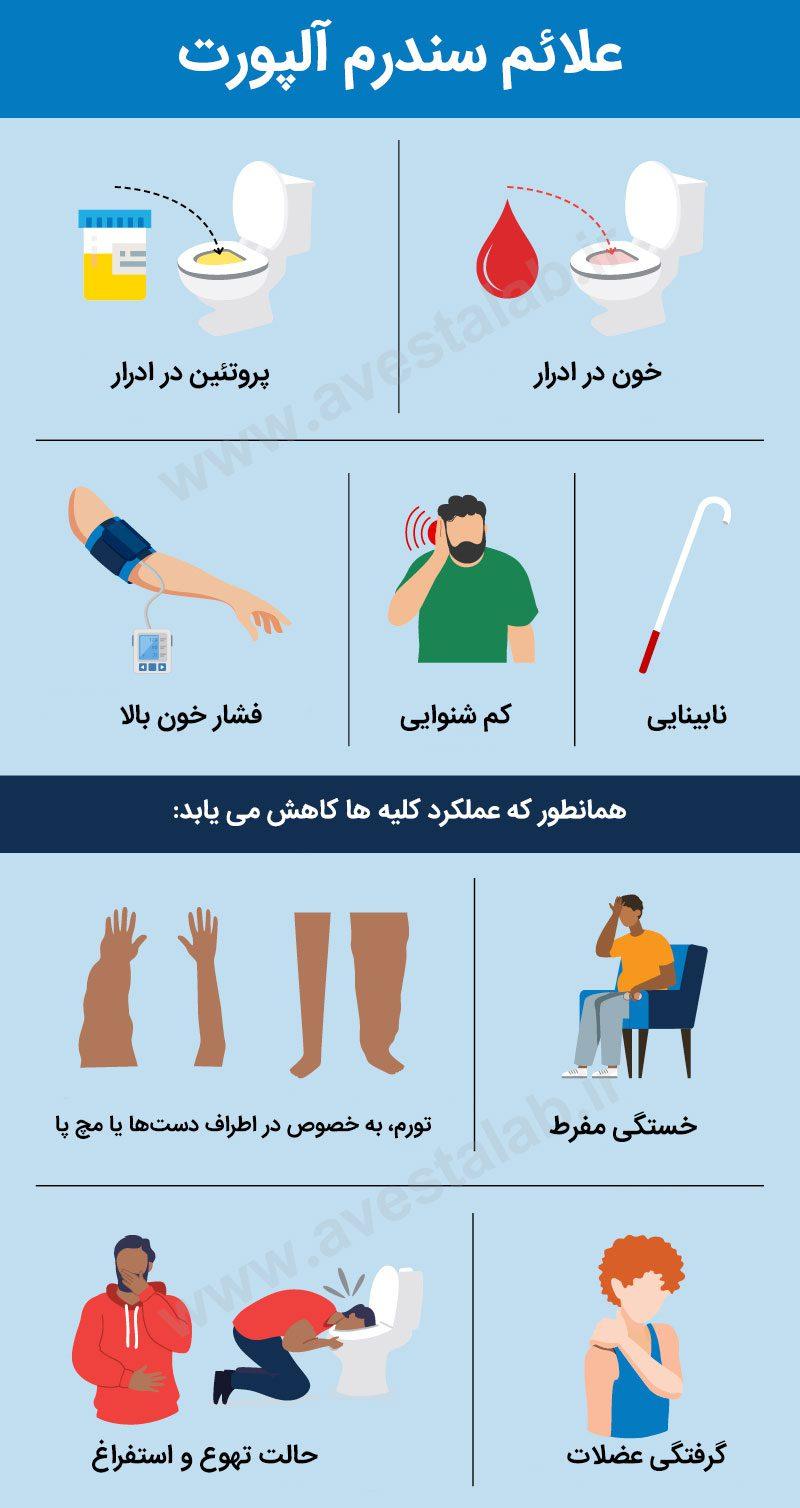
علائم بالینی اصلی سندرم آلپورت چیست؟
علائم سندرم آلپورت بسته به نوع بیماری، ممکن است متفاوت باشد که عبارتند از:
- وجود خون در ادرار که قابل مشاهده نیست (هماچوری میکروسکوپی).
- پروتئین در ادرار (پروتئینوری).
- بیماری مزمن کلیه یا نارسایی کلیه.
- کاهش شنوایی.
- مشکلات چشمی.
اولین علامت سندرم آلپورت، هماچوری میکروسکوپی است. هماچوری میکروسکوپی به این دلیل رخ می دهد که GBMهای غیرطبیعی، گلبول های قرمز خون را به ادرار سوق میدهند. افراد نمیتوانند سلول های خونی را با چشم غیرمسلح ببینند – فقط میتوان آن را زیر میکروسکوپ مشاهده نمود.
مردانی که XLAS دارند و هر کسی که ARAS دارد، در بدو تولد هماچوری میکروسکوپی خواهند داشت. اکثر زنانی که XLAS دارند به مرور زمان دچار هماچوری میکروسکوپی میشوند. همه افراد مبتلا به ADAS دچار هماچوری میکروسکوپی نمیشوند.
با شروع کاهش عملکرد کلیه، بیماری مزمن کلیه (CKD) ایجاد میشود. اکثر افراد تا زمانی که به نارسایی کلیه نرسند، علائم CKD ندارند.
علائم نارسایی کلیه عبارتند از:
- تورم، به خصوص در اطراف دستها یا مچ پا
- خستگی شدید
- حالت تهوع و استفراغ
- گرفتگی عضلات
کاهش شنوایی در مردانی که XLAS و افراد مبتلا به ARAS دارند، شایعتر است. با این حال، میتواند در هر کسی که مبتلا به سندرم آلپورت است، رخ دهد. کاهش شنوایی معمولاً تدریجی است. بسیاری از افراد تا زمانی که علائم شدیدتر نشود، متوجه کاهش شنوایی خود نمیشوند. اکثر افراد در شنیدن صداهای ریز مشکل دارند، اگرچه برخی افراد ممکن است در نهایت نتوانند هیچ صدایی را بشنوند. در نهایت ممکن است به سمعک نیاز داشته باشند. در موارد شدید، ناشنوایی رخ می دهد.
ممکن است طیف وسیعی از مشکلات در چشم ها ایجاد شود. برخی افراد بیشتر احتمال دارد سطح چشم خود را خراش دهند (ساییدگی قرنیه)، که بهبودی آن می تواند مدت زیادی طول بکشد. ساییدگی قرنیه ممکن است باعث آبریزش و درد چشم شود، اما معمولاً منجر به نابینایی نمی شود. برخی از افراد همچنین در قسمت شفاف چشم خود که به تمرکز بینایی آنها (عدسی) کمک میکند، دچار مشکل میشوند که در نهایت میتواند منجر به آب مروارید شود.
چه عواملی باعث سندرم آلپورت می شود؟
جهش در ژن های کلاژن باعث سندرم آلپورت می شود.
تشخیص و آزمایشها
در ابتدا پزشک علائم بیمار را بررسی کرده و در مورد سابقه خانوادگی بیولوژیکی او سوال می کند. آزمایش های مختلفی نیز میتوانند به پزشک در تشخیص سندرم آلپورت کمک کنند.
این آزمایشها عبارتند از:
آزمایش ادرار: آزمایش ادرار جنبه های بصری، شیمیایی و میکروسکوپی ادرار را بررسی می کند. این آزمایش میتواند به پزشک در شناسایی خون یا پروتئین در ادرار کمک کند.
آزمایش کلیرانس کراتینین یا آزمایش های خون سیستاتین C: این آزمایش ها سطح کراتینین و پروتئین سیستاتین C را در خون اندازهگیری می کنند. این آزمایشها نشان میدهند که کلیهها چقدر خوب خون را فیلتر میکنند.
میزان فیلتراسیون گلومرولی تخمینی (eGFR): در این آزمایش پزشک مقدار کراتینین یا سیستاتین C را محاسبه میکند. این مقدار تخمین می زند که کلیهها چقدر خوب خون را تصفیه میکنند.
بیوپسی کلیه: پزشک چند نمونهی کوچک از بافت کلیه را برداشته و برای بررسی دقیق زیر میکروسکوپ به آزمایشگاه میفرستد. این نمونهها میتوانند تغییرات ساختاری مختلفی را که عملکرد کلیه را تحت تأثیر قرار دادهاند، نمایان کنند.
در مبتلایان به سندرم آلپورت، غشای پایه گلومرولی (GBM) معمولاً نازکتر از حد طبیعی به نظر میرسد، اما در برخی نواحی ممکن است ضخیم و نامنظم شده باشد. در موارد پیشرفتهتر بیماری، ممکن است آسیب هایی مانند جای زخم (فیبروز) در واحدهای فیلترکننده کلیه و ساختارهای پشتیبان مشاهده شود.
آزمایش ژنتیک: آزمایش ژنتیک می تواند جهش در ژنهای کلاژن را شناسایی کند.
آزمایش شنوایی: پزشک در صورت مشکوک بودن به سندرم آلپورت، ممکن است آزمایش شنوایی (ادیوگرام) را تجویز کند. پزشکان باید برای هر کسی که سندرم آلپورت دارد، آزمایش شنوایی تجویز کنند. بیمار باید هر چند سال یکبار آزمایش شنوایی را برای نظارت بر شنوایی و مقدار شدت آن انجام دهد.
معاینه چشم: یک متخصص چشم پزشک بینایی بیمار را ارزیابی میکنند و سطح چشم (قرنیه)، عدسی و پشت چشم (شبکیه) را بررسی میکنند تا ببینند آیا سندرم آلپورت بر این نواحی تأثیر میگذارد یا خیر. آنها همچنین ممکن است آزمایش تصویربرداری توموگرافی انسجام نوری (OCT) انجام دهند.

درمان سندرم آلپورت
هیچ درمانی برای سندرم آلپورت وجود ندارد. با این حال، اکنون درمان هایی در تعبیه شدند که روند زوال کلیه را کند کرده و نارسایی کلیه را به تأخیر میاندازند.
پزشک ممکن است موارد زیر را تجویز کند:
مهارکنندههای آنزیم تبدیل کننده آنژیوتانسین (ACE): مهارکنندههای ACE فشار خون و پروتئین موجود در ادرار را کاهش می دهند و به محافظت از کلیهها کمک میکنند. مردانی که XLAS دارند یا هر کسی که ARAS دارد باید پس از تشخیص، مصرف مهارکنندههای ACE را شروع کند. زنانی که XLAS یا ADAS دارند، باید مهارکنندههای ACE را پس از شروع ظاهر شدن پروتئین در ادرار یا حتی در زمان تشخیص، شروع کنند.
مسدودکنندههای گیرنده آنژیوتانسین II (ARB): داروهای ARB مشابه مهارکنندههای ACE هستند و مزایای مشابهی دارند.
مهارکنندههای نوع 2 سدیم-گلوکز منتقلشده (SGLT-2): این داروها در صورت ابتلا به بیماری مزمن کلیه یا سندرم آلپورت به کاهش خطر نارسایی کلیه کمک میکنند. پزشک ممکن است این داروها را به مهارکننده ACE یا ARB اضافه کند. سازمان غذا و داروی آمریکا (FDA) همه مهارکنندههای SGLT-2 را برای کمک به درمان بیماری مزمن کلیه تأیید نکرده است. همچنین اگر میزان فیلتراسیون گلومرولی بیمار خیلی پایین باشد، پزشک آنها را تجویز نمیکند.
رژیم غذایی کنترلشده با سدیم: محدود کردن مقدار نمک و سدیم در رژیم غذایی به کاهش فشار خون کمک میکند و می تواند به حفظ سلامت کلیه و قلب کمک کند.
آیا پیوند کلیه می تواند سندرم آلپورت را درمان کند؟
بله و خیر. پیوند کلیه به فرد، کلیهای با کلاژن نوع IV طبیعی و غشاهای فیلترکننده میدهد. در نتیجه، سندرم آلپورت در کلیه جدید عود نخواهد کرد.
با این حال، پیوند کلیه به سایر علائم سندرم آلپورت، مانند کاهش شنوایی یا مشکلات چشمی، کمکی نخواهد کرد.
پیشگیری
شما نمیتوانید از سندرم آلپورت پیشگیری کنید، اما آگاهی از سابقه خانوادگی میتواند در تشخیص زودهنگام آن کمک کند. آگاهی زودهنگام همچنین می تواند به جلوگیری از انتقال آن به فرزند بیولوژیکی کمک کند.
تشخیص زودهنگام سندرم آلپورت و شروع درمان با مهارکنندههای ACE/ARB و مهارکنندههای SGLT-2 بهترین راه برای به تأخیر انداختن نارسایی کلیه است.
اگر پزشک در ادرار شما خون تشخیص دهد، ایده خوبی است که آزمایش های بیشتری برای سندرم آلپورت انجام دهید، به خصوص اگر مشکل شنوایی یا کاهش عملکرد کلیه دارید.
اگر سابقه خانوادگی هماچوری دارید، پزشک باید ادرار شما را از نظر خون، آزمایش کند و آزمایش خون برای بررسی عملکرد کلیه تجویز کند.
اقدامات احتیاطی
- مصرف مهارکنندههای ACE، ARBها یا مهارکنندههای SGLT-2 طبق تجویز پزشک
- اجتناب از مصرف داروهای ضدالتهاب غیراستروئیدی (NSAIDs)، که در صورت عملکرد غیرطبیعی کلیه یا سندرم آلپورت، ممکن است سرعت نارسایی کلیه را افزایش دهند.
- بررسی شنوایی
- مراقبت از سلامت روان
تغییرات سبک زندگی
کاهش میزان نمک در رژیم غذایی
اگر به بیماری مزمن کلیه مبتلا هستید، ممکن است به یک رژیم غذایی خاص نیاز داشته باشید. این رژیم غذایی ممکن است شامل مصرف پروتئین حیوانی کمتر یا در نظر گرفتن یک رژیم غذایی گیاهی، اجتناب از غذاهای سرشار از پتاسیم در صورت بالا بودن سطح پتاسیم خون و محدود کردن کامل میزان پروتئین در رژیم غذایی در صورت بالا بودن سطح فسفر خون یا هورمون پاراتیروئید (PTH) باشد.
داشتن یک سبک زندگی سالم برای قلب می تواند به کاهش خطر ابتلا به بیماری قلبی، دیابت و سایر بیماری هایی که ممکن است باعث نارسایی کلیه شوند، کمک کند. تمرینات سالم و حفظ وزن سالم برای قلب مانند پیادهروی سریع، دویدن، شنا، دوچرخهسواری و طناب زدن می تواند مفید باشد. همچنین اجتناب از سیگار و سایر محصولات دخانیاتی می تواند در این امر دخیل باشند.
مطالب مشابه: