سندرم پرادر-ویلی چیست؟ علت، علائم، تشخیص و درمان

سندرم پرادر-ویلی چیست؟ علت، علائم، تشخیص و درمان
سندرم پرادر-ویلی (Prader-Willi syndrome) یک بیماری ژنتیکی است که بسیاری از قسمت های بدن را تحت تاثیر قرار می دهد. در دوران نوزادی، این بیماری با ضعف تون عضلانی (هیپوتونی)، مشکلات تغذیه ای، رشد ضعیف و تاخیر در رشد مشخص می شود. از دوران کودکی، افراد مبتلا، دچار گرسنگی شدید می شوند که منجر به پرخوری مزمن (هیپرفاژی) و چاقی می شود. برخی از افراد مبتلا به سندرم پرادر-ویلی، به ویژه افراد چاق، به دیابت نوع 2 (شایع ترین نوع دیابت)، نیز مبتلا می شوند.
افراد مبتلا به سندرم پرادر-ویلی معمولاً دارای اختلال ذهنی خفیف تا متوسط و ناتوانی های یادگیری نیز هستند. مشکلات رفتاری از جمله طغیان های چشم، لجبازی و رفتارهای وسواسی مانند کندن پوست و ناهنجاری های خواب در چنین افرادی شایع است. از دیگر ویژگی های این بیماری می توان به ویژگی های متمایز صورت مانند پیشانی باریک، چشمان بادامی شکل و دهان مثلثی، قد کوتاه و دست ها و پاهای کوچک اشاره کرد. برخی از افراد مبتلا به سندرم پرادر-ویلی، پوست و موهای روشن غیرمعمولی دارند، هم مردان و هم زنان مبتلا اندام تناسلی توسعه نیافته ای دارند، بلوغ آنها با تاخیر و یا ناقص است و بیشتر آنها قادر به باروری نیستند.
علت سندرم پرادر-ویلی چیست؟
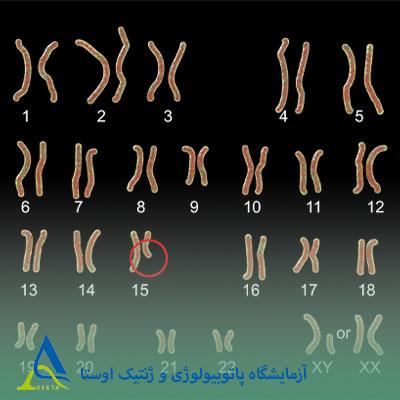
سندرم پرادر-ویلی یک اختلال ژنتیکی است که به دلیل از بین رفتن عملکرد برخی ژنها در ناحیه خاصی از کروموزوم 15 ایجاد میشود. هر فرد به طور طبیعی یک نسخه از این کروموزوم را از پدر و یک نسخه را از مادر به ارث میبرد. برخی ژنها تنها در نسخه پدری فعال هستند و نسخه مادری آنها غیرفعال باقی میماند؛ این فرآیند «نقشپذیری ژنومی» نام دارد.
در حدود 70% موارد، علت بروز این سندرم حذف بخشی از کروموزوم 15 پدری است. بنابراین، در این حالت، ژنهای حیاتی موجود در این ناحیه از بین میروند، زیرا نسخه پدری حذف شده و نسخه مادری نیز غیرفعال است. در حدود 25% دیگر، فرد هر دو نسخه کروموزوم 15 را از مادر دریافت میکند و نسخه پدری وجود ندارد. این وضعیت «دیزومی تکوالدی مادری» نام دارد. به ندرت، این سندرم میتواند به دلیل جابهجاییهای کروموزومی، تغییرات ژنتیکی یا عوامل دیگری ایجاد شود که باعث غیرفعال شدن غیرطبیعی ژنهای نسخه پدری میگردند.
ویژگی های اصلی سندرم پرادر-ویلی
ویژگیهای اصلی سندرم پرادر-ویلی ناشی از از دست رفتن عملکرد چندین ژن روی کروموزوم 15 است. برخی از این ژنها دستور ساخت مولکولهایی به نام «RNAهای کوچک هستهای» (snoRNA) را فراهم میکنند. این مولکولها وظایف مختلفی دارند، از جمله تنظیم فعالیت انواع دیگر RNAها که در ساخت پروتئینها و انجام فعالیتهای سلولی نقش مهمی ایفا میکنند. شواهد نشان میدهد که حذف خوشهای خاص از این ژنها به نام SNORD116 احتمالاً نقش مهمی در بروز علائم این بیماری دارد، هرچند مکانیزم دقیق آن هنوز مشخص نیست.
در بعضی از مبتلایان، حذف ژن OCA2 که در ناحیه حذفشده کروموزوم 15 قرار دارد، باعث روشنتر شدن غیرمعمول پوست و مو میشود. این ژن مسئول تولید پروتئینی است که در تعیین رنگ پوست، مو و چشم نقش دارد. با این حال، حذف آن تأثیری بر سایر علائم اصلی سندرم پرادر-ویلی ندارد.
پژوهشگران همچنان در حال بررسی ژنهای دیگر موجود روی کروموزوم 15 هستند که ممکن است در بروز علائم و نشانههای اصلی این اختلال نقش داشته باشند.
ژن OCA2
ژن OCA2 (که قبلاً ژن P نامیده میشد) دستورالعملهایی برای ساخت پروتئینی به نام پروتئین P ارائه میدهد. این پروتئین در ملانوسیتها قرار دارد، که سلولهای تخصصی هستند که رنگدانهای به نام ملانین تولید میکنند. ملانین مادهای است که به پوست، مو و چشمها رنگ میدهد. ملانین همچنین در بافت حساس به نور در پشت چشم (شبکیه) یافت میشود، جایی که در بینایی طبیعی نقش دارد.
اگرچه عملکرد دقیق پروتئین P ناشناخته است، اما برای رنگدانهسازی طبیعی ضروری است و احتمالاً در تولید ملانین نقش دارد. در ملانوسیتها، پروتئین P ممکن است مولکولها را به داخل و خارج از ساختارهایی به نام ملانوزوم (جایی که ملانین تولید میشود) منتقل کند. محققان معتقدند که این پروتئین همچنین ممکن است به تنظیم اسیدیته نسبی (pH) ملانوزومها کمک کند. کنترل دقیق pH برای اکثر فرآیندهای بیولوژیکی ضروری است.
وراثت
بیشتر موارد سندرم پرادر-ویلی ماهیت ارثی ندارند، بهویژه آنهایی که ناشی از حذف بخشی از کروموزوم 15 پدری یا «دیزومی تکوالدی مادری» هستند. این تغییرات ژنتیکی معمولاً بهصورت تصادفی و در حین تشکیل سلولهای جنسی (تخمک یا اسپرم) یا در مراحل ابتدایی رشد جنین رخ میدهند. به همین دلیل، اغلب افراد مبتلا سابقهای از این بیماری در خانواده خود ندارند.
در موارد نادر، تغییر ژنتیکی مرتبط با سندرم پرادر-ویلی میتواند به صورت ارثی منتقل شود. برای مثال، گاهی ممکن است تغییری که باعث غیر فعال شدن غیرطبیعی ژنهای نسخه پدری کروموزوم 15 میشود، از یک نسل به نسل دیگر به ارث برسد.
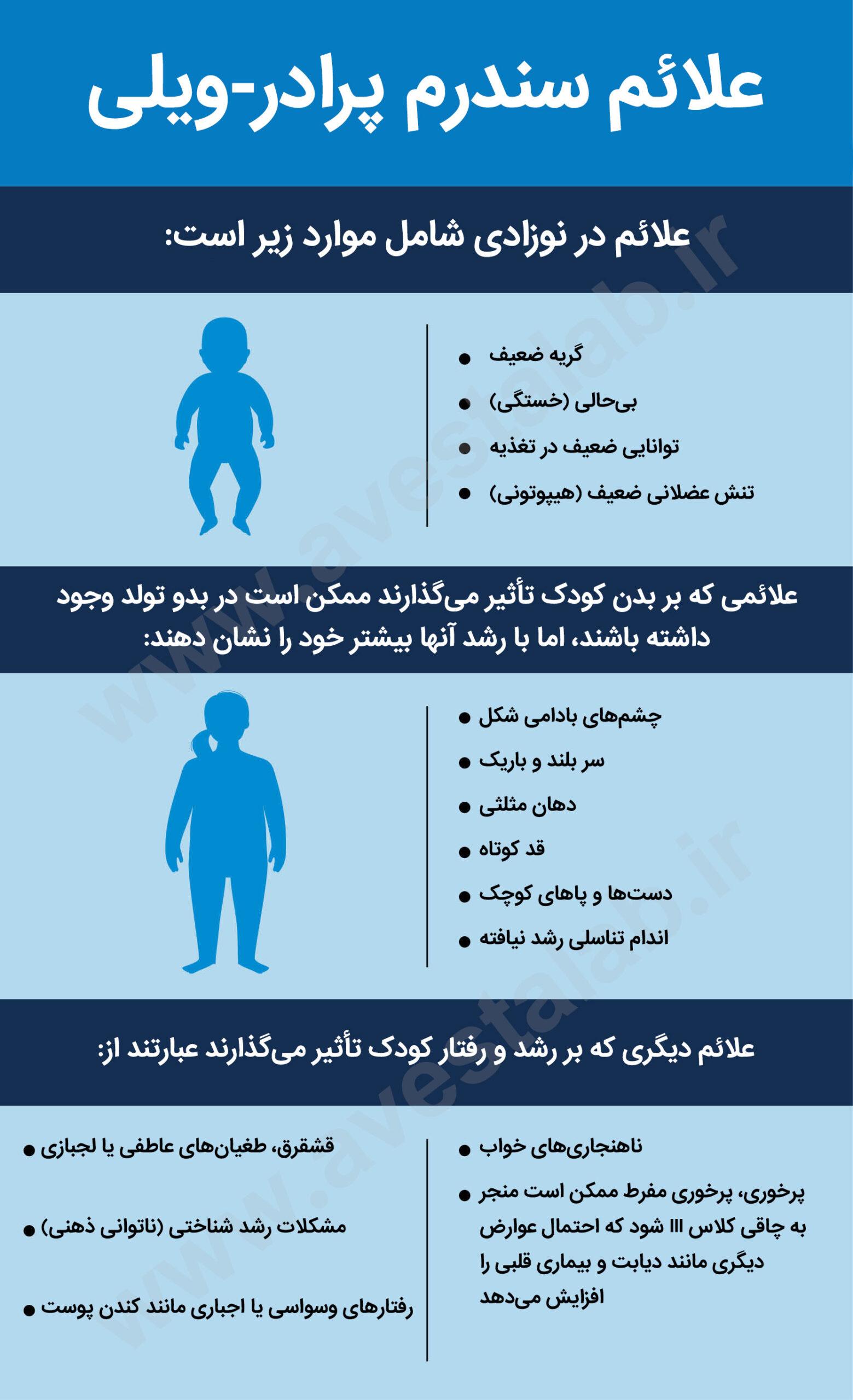
علائم سندرم پرادر-ویلی چیست؟
سندرم پرادر-ویلی هر فرد را به طور متفاوتی تحت تأثیر قرار میدهد.
علائمی که در دوران نوزادی ظاهر میشوند ممکن است شامل موارد زیر باشند:
- گریه ضعیف
- بیحالی (خستگی)
- توانایی ضعیف در تغذیه
- تنش عضلانی ضعیف (هیپوتونی)
علائمی که بر بدن کودک تأثیر میگذارند ممکن است در بدو تولد وجود داشته باشند، اما با رشد آنها بیشتر خود را نشان دهند. این علائم ممکن است شامل موارد زیر باشد:
- چشمهای بادامی شکل
- سر بلند و باریک
- دهان مثلثی
- قد کوتاه
- دستها و پاهای کوچک
- اندام تناسلی رشد نیافته
علائم دیگری که بر رشد و رفتار کودک تأثیر میگذارند عبارتند از:
- قشقرق، طغیانهای عاطفی یا لجبازی
- مشکلات رشد شناختی (ناتوانی ذهنی)
- رفتارهای وسواسی یا اجباری مانند کندن پوست
- ناهنجاریهای خواب
- پرخوری، پرخوری مفرط ممکن است منجر به چاقی کلاس III شود که احتمال عوارض دیگری مانند دیابت و بیماری قلبی را افزایش میدهد.
عوارض مرتبط با سندرم پرادر-ویلی چیست؟
بسیاری از افراد مبتلا به سندرم پرادر-ویلی به دلیل پرخوری دچار چاقی میشوند. عوارض ناشی از چاقی ممکن است شامل موارد زیر باشد:
- مشکلات قلبی
- دیابت (نوع 2)
- فشار خون بالا
- مشکلات تنفسی
- آپنه خواب (قطع تنفس در طول خواب)
چاقی یک بیماری پیچیده اما قابل کنترل است. پزشک میتواند در مورد چگونگی کمک به کودک و جلوگیری از عوارض، توصیه هایی ارائه دهد.
تشخیص و آزمایشها
پزشک پس از معاینه فیزیکی و آزمایشهای ژنتیکی، سندرم پرادر-ویلی را تشخیص میدهد. پزشک در طول معاینه به دنبال علائم فیزیکی این بیماری میگردد و در مورد علائم کودک، از جمله عادات غذایی و رفتار او، از والدین سؤال میکند. اگر پزشک به سندرم پرادر-ویلی مشکوک باشد، آزمایش ژنتیک انجام میدهد که یک آزمایش خون است که ناهنجاریهای ناشی از تغییرات در DNA کودک را تشخیص میدهد.
آزمایشات تکمیلی
آزمایش متیلاسیون DNA (DNA Methylation Analysis)
دقیقترین روش برای تشخیص سندرم پرادر-ویلی، حتی زمانی که نوع تغییر ژنتیکی مشخص نیست.
این تست میتواند تعیین کند که آیا ژنهای نسخه پدری در ناحیه مورد نظر کروموزوم 15 فعال هستند یا خیر.
آنالیز کاریوتایپ (Karyotyping)
بررسی ساختار کروموزومها برای شناسایی حذفها، جابهجاییها یا سایر ناهنجاریهای ساختاری در کروموزوم 15.
آزمایش FISH (Fluorescence In Situ Hybridization)
یک روش دقیق برای شناسایی حذف بخشی از کروموزوم 15 پدری که در بیشتر بیماران مشاهده میشود.
آزمایش Array CGH یا Microarray
بررسی کل ژنوم برای شناسایی حذفها یا افزودههای کوچک در DNA که با روشهای معمول قابل تشخیص نیستند.
بررسی دیزومی تکوالدی (Uniparental Disomy Test)
تشخیص اینکه آیا فرد هر دو نسخه کروموزوم 15 را از مادر به ارث برده است یا خیر.
تست توالییابی ژنها (Gene Sequencing)
در موارد نادر، برای شناسایی جهشهای کوچک یا تغییرات در ژنهای خاصی که ممکن است در بروز بیماری نقش داشته باشند.
آزمایشهای حمایتی و ارزیابیهای تکمیلی
آزمایشهای هورمونی: برای بررسی سطح هورمون رشد، هورمونهای تیروئید و عملکرد غدد جنسی.
ارزیابی تغذیهای و متابولیک: جهت بررسی چاقی، مقاومت به انسولین یا مشکلات متابولیکی.
آزمایش خواب: برای شناسایی آپنه یا سایر اختلالات خواب که در این بیماران شایع است.
درمان سندرم پرادر-ویلی
درمان سندرم پرادر-ویلی بر کنترل علائم و پیشگیری از عوارض تمرکز دارد که میتواند شامل موارد زیر باشد:
دستگاههایی مانند سر شیشههای مخصوص، برای کمک به نوزادان برای دریافت تغذیه کافی.
کمک به کودک برای تغذیه مناسب، از جمله رژیم غذایی کم کالری و کنترل میزان غذای او.
داروهایی برای افزایش مقدار هورمونهای خاص، مانند هورمون رشد، و تستوسترون یا گنادوتروپین جفتی انسان (HCG) برای پسران و استروژن برای دختران.
درمانهای حمایتی مانند فیزیوتراپی، گفتاردرمانی و آموزش ویژه برای بهبود عملکرد جسمی و شناختی.
پیشگیری
سندرم پرادر-ویلی یک بیماری ژنتیکی است و راهی برای پیشگیری کامل از آن وجود ندارد. در بسیاری از موارد، این اختلال ناشی از یک تغییر ژنتیکی تصادفی و غیرقابل پیشبینی است که ارتباطی با اقدامات والدین پیش یا در طول بارداری ندارد.
برای آگاهی بیشتر از احتمال ابتلای فرزند به بیماریهای ژنتیکی، میتوانید با پزشک یا مشاور ژنتیک مشورت کرده و انجام آزمایشهای ژنتیکی را مدنظر قرار دهید.
نحوه تعیین وقت مشاوره در آزمایشگاه پاتوبیولوژی اوستا
حضوری: با مراجعه به آزمایشگاه پاتوبیولوژی اوستا از خدمات مشاوره ژنتیک بهرهمند شوید.
آدرس: خیابان ولیعصر – خیابان مطهری – نبش خیابان لارستان – پلاک 414، تهران
شماره تماس: 02188801071 – 02188802408 – avesta_lab@
ساعات کاری: شنبه تا پنجشنبه، 7:00 الی 19:00
مطالب مشابه: